 |
 |
|
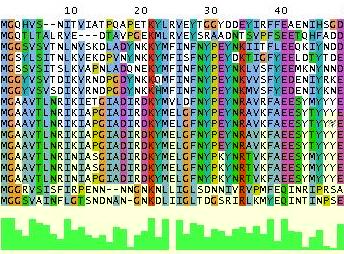
Multiple alignments of protein sequences are an important tool for studying proteins, because
they allow identification of conserved amino acid sites. This is very useful
in designing experiments to test the function of particular amino acid sites;
in predicting the structure of proteins;
and in identifying new members of protein families.
|  |
Our laboratory collaborates on the widely used Clustal series of programs for multiple sequence alignment. These are maintained as collaboration with Toby Gibson (EMBL, Heidelberg) and Julie Thompson (ICGEB, Strasbourg). Clustal is a free package that can rapidly and simply align hundreds of nucleic acid or amino acid sequences. It runs on PCs, MACs, Linux and Unix machines and can be downloaded for free or you can run it remotely using the WWW.
T-Coffee is a package for carrying out very accurate multiple sequence alignments and is capable of exploiting data from a mixture of sources such as primary sequences and 3-D structures. It is being developed and maintained by Cedric Notredame in Marseilles and is freely available and will run on Unix machines or well-endowed PCs. You can download it or run it using the WWW. Our laboratory collaborates with Dr. Notredame in developing methods for using T-Coffee with structural information. For more information on T-Coffee, see the T-Coffee website.